A Theoretical Study: Solvent Effects on the Electronic Excitations of (N,N)-bridged
4-Aminobenzonitriles
Rudolf Schamschule1,+, Andreas B. J. Parusel1,
Gottfried Köhler1,2
1 Institut für Theoretische Chemie und Strahlenchemie,
University of Vienna, Althanstr. 14, 1090 Vienna, Austria.
2 Austrian Society for Aerospace Medicine - Institute for Space Biophysics,
Lustkandlgasse 52, 1090 Vienna, Austria. + Corresponding author.
First presented at the first internet conference on
photochemistry and photobiology
Keywords: Aminobenzonitriles, Charge transfer, Dual fluorescence, Solvent Effects
Contents
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Results for N-azetidinyl-benzonitrile
(4BN)
3.2. Results for N-pyrolidinyl-benzonitrile
(PYRBN)
3.3. Results for N-piperidinyl-benzonitrile
(PIPBN)
3.4. Comparison to Experimental Data
4. Conclusion
5. Acknowledgements
6. References
N-azetidinyl-benzonitrile (4BN), N-pyrolidinyl-benzonitrile (PYRBN) and
N-piperidinyl-benzonitrile (PIPBN)
Abstract
The influence of polar environments on the excited state properties is investigated for
a series of four to six membered N,N-bridged 4-aminobenzonitriles. The self
consistent reaction field model (SCRF), with an extension for the description of solvent
effects on the electronically excited states, is used in a semiempirical AM1 based method.
The rotational isomerization of the alkyl-amino group, a pseudo Jahn-Teller mechanism, and
the rehybridization at the cyano carbon are investigated as possible explanations for the
observed occurrence of dual fluorescence. Based on these solvent calculations,
fluorescence wavelengths are compared to experimental data from literature. The RICT
(Rehybridization by Intramolecular Charge Transfer) and WICT (Wagging Intramolecular
Charge Transfer) models are rejected as explanations of the dual fluorescence phenomenon
for this class of donor-acceptor compounds. The TICT (Twisted Intramolecular Charge
Transfer) is found to explain the dual fluorescence of each compound for itself. A
sufficiently small energy gap is found between the S1 and S2 excited
states, making vibronic coupling, as proposed by the pseudo Jahn-Teller hypothesis, an
accurate hypothesis for the description of the dual fluorescence behaviour of these
compounds.
back to Contents.
1. Introduction:
The phenomenon of dual fluorescence has been widely studied for 4-N,N-Dimethylamino-benzonitrile
(DMABN) and related electron donor-acceptor compounds. Besides the so-called normal
fluorescence out of the locally excited S1 state, a second, strongly
red-shifted emission can be observed with increasing solvent polarity. Since discovery of
this effect DMABN and derivatives by Lippert et al. [1], the strong polarity dependence of
the spectral position of the second, red-shifted fluorescence has been well documented
[2-5]. The increased quantum yield, when hydrocarbons are replaced by polar solvents
[6,7], combined with the dependence on environment polarity, indicate a highly dipolar
excited state achievable only by a charge transfer in combination with an internal
geometric relaxation out of the locally excited (LE) S1 state, and not
reachable by direct excitation.
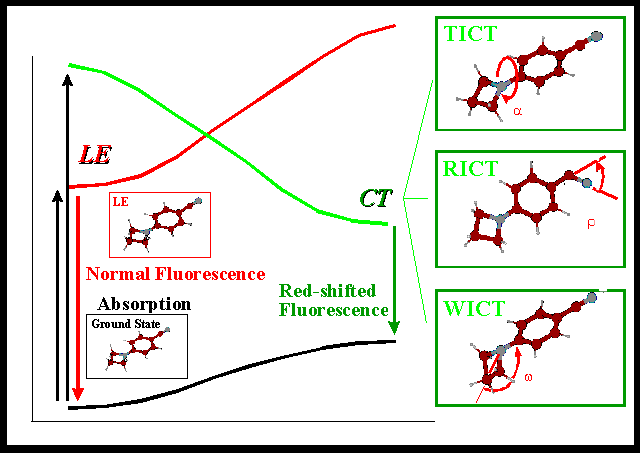
Figure 1a: Definition of the wagging angle  , the twist
angle , the twist
angle  , and the rehybridization angle , and the rehybridization angle 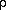 in
combination with the explanation of the WICT, TICT, and RICT hypotheses for 4BN. The
definitions for the other compounds are analogous. in
combination with the explanation of the WICT, TICT, and RICT hypotheses for 4BN. The
definitions for the other compounds are analogous.
The wagging angle , defining the pyramidalization of the
amino-group, and the twist angle , defining its out of the
benzonitrile plane rotation, as well as the rehybridization angle representing
the structural change of the molecules during the rehybridization of the cyano carbon are
presented for 4BN in Figure 1a. The definitions of these angles are analogous for the
other molecules.
The widely accepted TICT (Twisted Intramolecular Charge Transfer) hypothesis [2, 6-8]
predicts the intramolecular relaxation of the planar vertically excited state (LE for
locally excited) to a highly dipolar charge transfer state with perpendicular orientation
of the amino (electron donor) and benzonitrile (electron acceptor) moieties as the cause
for to the occurrence of the strongly redshifted fluorescence band. For the investigation
of this TICT model, the change of dipole moment and excitation energy of the relevant
electronically excited states are studied with increasing torsion angle of the amino group. In this TICT hypothesis, the twisting motion of the
amino group (see Figure 1a) causes a maximized charge separation, by means of a mutually
perpendicular conformation of the amino-donor and the benzonitrile-acceptor subunits. In
this charge transfer state, an electron is transferred from the nitrogen lone pair into
the unoccupied aromatic benzonitrile orbital.
The WICT model (Wagging Intramolecular Charge Transfer), proposed by Gorse and Pesquer
[9,10], predicts a charge transfer state, where the amino nitrogen lone pair orbital is
decoupled from the benzonitrile  -orbitals by means of an amino wagging
mode. Therefore the WICT model demands a strong deviation from planarity corresponding to
large values for the wagging angle . This change of the
pyramidalization of the amino nitrogen is the considered to be the sole relaxation mode
responsible for the dual fluorescence. With the wagging angle as its
characterizing coordinate, a change of the amino nitrogen from planar sp2 to
pyramidal sp3 hybridization is described as another possible hypothesis for the
description of the observed dual fluorescence phenomenon. -orbitals by means of an amino wagging
mode. Therefore the WICT model demands a strong deviation from planarity corresponding to
large values for the wagging angle . This change of the
pyramidalization of the amino nitrogen is the considered to be the sole relaxation mode
responsible for the dual fluorescence. With the wagging angle as its
characterizing coordinate, a change of the amino nitrogen from planar sp2 to
pyramidal sp3 hybridization is described as another possible hypothesis for the
description of the observed dual fluorescence phenomenon.
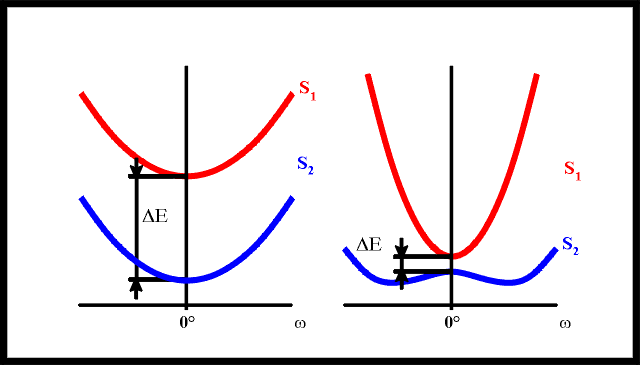
Figure 1b: Energy diagram of the two lowest excited states. According to the
pseudo Jahn Teller hypothesis, a sufficient small energy gap  E is
necessary for the occurrence of dual fluorescence. With a large energy gap between the S1
and the S2 state (left), no vibronic coupling occurs. With a sufficiently small
energy gap (right), the characteristic double minima of the S1 state is
observed, resulting in vibronic coupling between these two states. E is
necessary for the occurrence of dual fluorescence. With a large energy gap between the S1
and the S2 state (left), no vibronic coupling occurs. With a sufficiently small
energy gap (right), the characteristic double minima of the S1 state is
observed, resulting in vibronic coupling between these two states.
Vibronic coupling can be achieved by a sufficiently small energy gap between two
interacting states S1 and S2 (see Figure 1b). I n combination with
the presence of a promoting mode, such as nitrogen inversion, this coupling can result in
a charge transfer state leading to the second fluorescence band. This so-called solvent
induced pseudo Jahn-Teller effect model [4, 11-14] was first introduced by Zachariasse et
al. Contrary to the WICT hypothesis, the amino wagging angle only varies between the
ground state minima at =-35o through the planar
conformation (at =0o to =35o
In 1996, an in-plane bending of the cyano group was calculated [15-17] as another
possible main relaxation coordinate (see Figure 1a). With this so-called RICT hypothesis
(Rehybridization by Intramolecular Charge Transfer), a sp to sp2
rehybridization of the carbon atom of the cyano group, causing a relaxation by bending of
the cyano angle, is presented as the main stabilizing factor for the formation of the
charge transfer state.
Scheme 1: Molecular structures of 4BN, PYRBN and PIPBN (left, middle and right).
Rate constants for PYRBN and PIPBN measured with laser techniques were presented by W.
Rettig [18]. Further studies on the solvent dependent dynamics of the formation of the
TICT state at different temperatures are known from literature for PYRBN and PIPBN [19].
To determine the excitation energy dependence of the fluorescence, low temperature studies
in n-propanol were discussed by D. Braun and W. Rettig for PYRBN [20]. In 1994, the dual
fluorescence was investigated by K. A. Zachariasse et al. [11], leading to the proposal of
the pseudo Jahn Teller effect as a cause of the dual fluorescence phenomenon. The solvent
polarity dependence of the dual fluorescence was measured for a series of three- to
eight-membered (N,N)-bridged 4-Aminobenzonitriles [21].
In a recent paper, we reported on semiempirical electronic structure calculations in
gas phase, describing the excited state potential energy surfaces of
N-azetidinyl-benzonitrile (4BN), N-pyrolidinyl-benzonitrile (PYRBN) and
N-piperidinyl-benzonitrile (PIPBN) [22] (see Scheme 1). Different possible structural
relaxation modes were compared for these N,N-bridged aminobenzonitriles.
Distinctive charge separation and large dipole moments were correlated to the occurrence
of the red-shifted fluorescence. Based on these gas phase calculations, we rejected the
WICT model as an explanation of the dual fluorescence phenomenon. The RICT model yielded
high energy barriers against the cyano bending mode, but due to the resulting stabilized
charge transfer states, the gas phase calculations were insufficient for a definite
rejection of this RICT hypothesis. The TICT and the pseudo Jahn-Teller hypotheses were
found as possible concurring relaxation modes for this class of donor-acceptor molecules.
Based on the gas phase calculations presented in the previous paper, the effects of a
polar solvent on the excited state properties are explicitly calculated for these
molecules. The SCRF (self consistent reaction field) model [23] is used for the
description of these solvent effects. Solvated excited state properties of
N-azetidinyl-benzonitrile (4BN), N-pyrolidinyl-benzonitrile (PYRBN) and
N-piperidinyl-benzonitrile (PIPBN) are presented as a means of describing the
spectroscopic behavior of these compounds. To model the solvent polarity dependence of the
fluorescence shift, calculated fluorescence wavelengths for acetonitrile and diethylether
are compared to experimental data.
back to Contents.
2. Computational Methods:
In a previous paper, gas phase potential energy surface scans along internal relaxation
coordinates were presented for these N-bridged aminobenzonitriles [22]. Based on these gas
phase calculations performed with the semiempirical AM1 method [24], the effects of
increasing solvent polarity are calculated with the SCRF (self consistent reaction field)
[21] approach. In the SCRF model for ground and excited states, the solvent is treated as
a polarizable dielectric continuum, characterized by its refractive index n and its
dielectric constant  . The so called reaction field, an electrostatic
field, is generated by the polarization of the continuum. The polarization of the solute
within the solvent cavity, itself influencing the field, is brought to self consistence
within a self consistent field (SCF) iteration sequence. The solvated excited states are
described by configuration interaction calculations with the CISD method with 10 active
orbitals including all single and double excitations. Excitation energies and orbital
analysis as well as dipole moments are used as a means of characterizing the properties of
the relevant charge transfer states along the relaxation coordinates. . The so called reaction field, an electrostatic
field, is generated by the polarization of the continuum. The polarization of the solute
within the solvent cavity, itself influencing the field, is brought to self consistence
within a self consistent field (SCF) iteration sequence. The solvated excited states are
described by configuration interaction calculations with the CISD method with 10 active
orbitals including all single and double excitations. Excitation energies and orbital
analysis as well as dipole moments are used as a means of characterizing the properties of
the relevant charge transfer states along the relaxation coordinates.
The torsion angle and the wagging angle of
the amino moiety are presented as possible relaxation modes resulting in a red-shifted
fluorescence band. The torsion and wagging angles, and , respectively, were varied in steps of 10o with the values of
the twist angle ranging from 0o (planar conformation) to
90o (twisted conformation). The wagging angle was scanned
from 0o with a planar sp2 conformation up to 70o with a
sp3 hybridized amino nitrogen. Due to the absence of a sufficiently stabilized
charge transfer state for large wagging angles , the potential energy
scans for varying wagging angles are only briefly discussed to reject of the WICT model
for these compounds. Additionally, the cyano bending angle is scanned
from 180o to 110o. For a correct description of the changing
hybridization of the cyano carbon, the Ccyano- Ncyano bond distance
was varied linear with the cyano bending angle, as done by Domcke and Sobolewski [15]. The
bond length varied from 1.14 Ĺ for =180o (sp hybridization,
corresponding to a CN triple bond), to 1.30 Ĺ for =120o (sp2
hybridization, CN double bond), and up to 1.33 Ĺ for =110o.
For all computations, the semiempirical AM1 Hamiltonian included in the VAMP program
package, version 6.1 [25], is used. These calculations were performed on Indy workstations
(MIPS R4400, Silicon Graphics Inc.).
back to Contents.
Results and Discussion:
In a previous publication, relaxed scans of the gas phase potential energy surface of
the electronic ground state were used to illustrate the change in the orbital character as
well as changes in the energies of the excited states along certain relaxation coordinates
[22]. While gas phase calculations are a fairly reliable tool for the qualitative
description of these electronic excitation processes, only explicit inclusion of solvent
effects can lead to a quantitative description of energy barriers and solvent
stabilization effects. The excited state energies are investigated in acetonitrile for
selected excited states of interest for the description of the dual fluorescence
phenomenon.
back to Contents.
3.1. Results for N-azetidinyl-benzonitrile:
The excitation energies and orbital compositions of the charge transfer states
considered by the TICT (alkylamino rotation) and RICT (bending of the cyano group)
hypotheses are presented for 4BN in Figure 2.
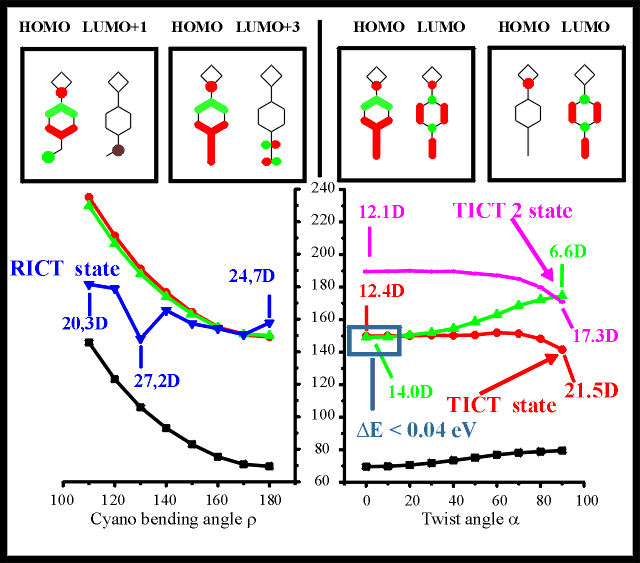
Figure 2: Excited state energies and orbital analyses for 4BN in acetonitrile
for varying cyano bending angle (bottom left) and for varying amino
twist angle (bottom right). Orbital compositions are presented (top,
from left to right) for the RICT state at =120o and for the
S3 (the charge transfer state becoming the RICT state upon cyano bending) at =180o and for the S2 charge transfer state at =0o and for the same state (TICT state) at =90o
For the twisting relaxation mode, the lowest two excited states are of special
interest. The S1 state is of B1 symmetry, consisting mainly of a
benzonitrile * transition. The S2
state, where an electron is transferred from the amino nitrogen lone pair to the
benzonitrile moiety, is a HOMO to LUMO charge transfer state of A symmetry. A change in
the character of the relevant orbitals towards increasing charge separation can be
observed for growing twist angles. For =90o, a
localization of the HOMO on the nitrogen of the amino donor group can be observed, with a
delocalization of the LUMO over the benzonitrile acceptor group. At about 20o
amino twisting, the energies of these two states cross, the HOMO to LUMO state (TICT
state) is lowered below the * state, which
in turn becomes the S2 state for twisted conformations. The twisting of the
amino group yields a strong diminished dipole moment for the * state, from 14.0 D for the untwisted conformation to 6.6D for
the twisted conformation. The dipole moment of the namino*
charge transfer state is increased from 12.4D (untwisted conformation) up to a dipole
moment of 21.5D for the twisted conformation (TICT state). In addition to these two
states, a higher excited state is stabilized by twisting of the amino group. Due to the
comparable properties (upon twisting of the alkylamino group) of this state and the TICT
state, this second, energetically higher charge transfer state is referred to as TICT2
state. Due to the stabilizing effects of the polar solvent, the S2 excited
state is lowered close to the S1 even for the untwisted (LE for locally
excited) conformation of 4BN, with an energy difference of only 0.04 eV, which is almost
isoenergetic. For increasing twist angles of the amino group, the high dipole moment of
the solvated TICT charge transfer state results in a strong stabilization of this excited
state. The energy barrier against the relaxation along this twisting mode is found to be
only 0.02 eV (see Table 1), which is surmountable by vibronic excitation at room
temperature, resulting in an unhindered twisting motion upon electronic excitation. This
small barrier facilitates the relaxation along the twisting mode, strongly supporting the
TICT mechanism as a possible description of the dual fluorescence phenomenon for 4BN.
Another dipolar charge transfer excited state, the TICT2 state, is strongly stabilized for
large twisting angles, with its energy decreased below the lowest * excited state for 90o twisting.
The inversion angle was considered as another possible relaxation
mode leading to an internal charge transfer state yielding the second, red shifted
fluorescence. In previous gas phase calculations, it was found that, due to the absence of
an energy stabilization, the wagging mode of the alkylamino group is unlikely to cause the
dual fluorescence. Even with the explicit inclusion of the effects of a polar solvent and
for large wagging angles ( > 60), intramolecular relaxation of
these molecules along the inversion mode leads to increasing excited state energy with
increasing wagging angle, with a drastic increase in the energy for angles greater than 50o.
The absence (for all three systems) of a stabilization of any charge transfer state below
the S1 state makes the WICT hypothesis unsuitable for all of the discussed N,N-bridged
aminobenzonitriles. Due to this rejection of the WICT hypothesis for this class of
molecules, this model is not further discussed in this paper.
The RICT hypothesis proposes the cyano bending mode, where the hybridization of the
cyano carbon atom from sp to sp2 is the sole relaxation mode responsible for
the occurrence of dual fluorescence. Contrary to the TICT mode, where the energy of the S2
state is decreased below energy of the former S1 state, a higher state with B1
symmetry (the so-called RICT state) is lowered in energy. Due to the high dipole moment of
this charge transfer state, this excited state is lowered from the S6 state in
gas phase [22] to the S3 in acetonitrile. Even though the dipole moment varies
only from 24.7 D for the unbent conformation, up to 27.2D for the energy minimum at =130o this state is drastically lowered in energy (below the
energy of the S1 state of the untwisted conformation). For larger angles, the
dipole moment rises again, to 20.3D for =110o accompanied by
a corresponding increase of the excited state energy. Even though the energy lowering of
this charge transfer state is in favor of this hypothesis, the prohibitively high energy
barrier of 0.71 eV makes the intramolecular relaxation along the cyano bending mode
impossible. The energy barriers against the relaxation along the twisting and cyano
bending modes are presented in Table 1.
| |
TICT |
RICT |
| 4BN |
0.02 eV |
0.71 eV |
| PYRBN |
0.12 eV |
0.14 eV |
| PIPBN |
No Barrier |
(No Minimum) |
There is no energy stabilization caused by the relaxation along the amino wagging mode.
Therefore no energy barriers are presented for the WICT model. For the cyano bending mode
in PIPBN, there is only a small barrier against the cyano bending mode. Nevertheless, the
RICT model does not offer a correct hypothesis for PIPBN, due to fact that the energy of
the amino-cyano charge transfer state is not lowered below the energy of the locally
excited (LE) state.
Table 2: Energy gap <E [eV] between the lowest excited states for the untwisted
conformation of 4BN, PYRBN and PIPBN.
| |
E (S2-S1) |
E (S3-S2) |
| 4BN |
0.04 |
0.76 |
| PYRBN |
0.03 |
0.73 |
| PIPBN |
0.05 |
0.03 |
The solvent induced pseudo Jahn Teller theory has been widely discussed as an
alternative hypothesis for the description of the dual fluorescence phenomenon for 4-(N,N-
Dimethylamino)-benzonitrile (DMABN). For 4BN, the explicit inclusion of solvent effects
(calculated in acetonitrile) results in an energy gap between the S1 and the S2
states of less than 0.04 eV. This small energy difference permits vibronic coupling
between these two states. Combined with the nitrogen inversion as a promoting mode, this
pseudo Jahn-Teller hypothesis is found to be a second possible explanation for the
emission of the red-shifted fluorescence.
The barrier for the amino twisting mode for 4BN implies the reduction of the normal
fluorescence band to small intensity in the spectrum of 4BN. This is found to be in
excellent agreement with experimental data [21], which shows an LE transfer band combined
with a smaller charge transfer emission.
back to Contents.
3.2. Results for N-pyrolidinyl-benzonitrile (PYRBN):
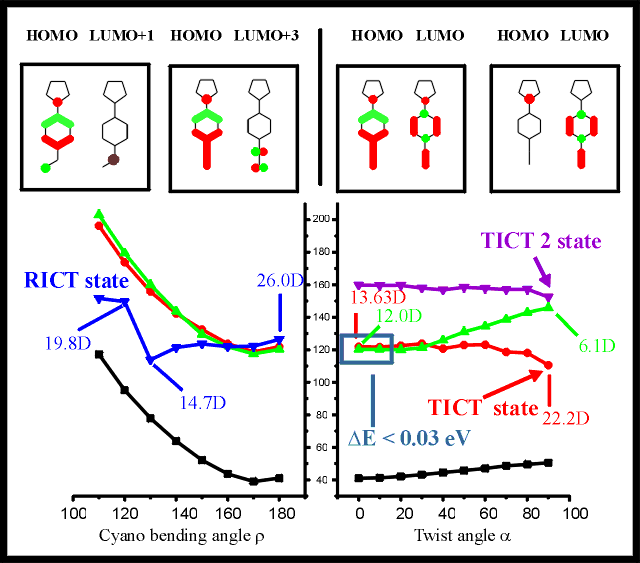
Figure 3: Excited state energies and orbital analyses for PYRBN in acetonitrile
for varying cyano bending angle (bottom left) and for varying amino
twist angle (bottom right). Orbital compositions are presented (top,
from left to right) for the RICT state at =120o and for the
S3 (the charge transfer state becoming the RICT state upon cyano bending) at =180o and for the S2 charge transfer state at =0o and for the same state (TICT state) at =90o
For PYRBN, the excitation energies and orbital compositions are presented in Figure 3
for varying rotation angles and cyano bending angles . For this five-membered N,N-bridged aminobenzonitrile, the lowest
two excited states are of special interest for the twisting relaxation mode. The S1
state is of found to be of B1 symmetry, consisting mainly of a benzonitrile * transition. For the S2 state an
electron is transferred from the amino nitrogen lone pair to the benzonitrile moiety
resulting in this HOMO to LUMO charge transfer state of A symmetry. A change in the
character of the relevant orbitals towards increasing charge separation can be observed
for larger values of . For the twisted conformation ( =90o), a strong localization of the HOMO orbital on the
nitrogen of the amino donor group can be observed, with a delocalized of the LUMO over the
benzonitrile acceptor group. The energy crossing of the charge transfer state below the * state is found between 30o and 40o
twist angle. For the conformation with the twisted amino group, a strongly diminished
dipole moment of 6.1 D is found for the *
state, decreased from 14.0 D for the conformation with the untwisted amino subunit. For
increasing twist angles of the amino group, the high dipole moment of the solvated TICT
charge transfer state results in a strong stabilization of this excited state. The dipole
moment of the namino* charge transfer state
increases from 12.0 D found for the untwisted conformation up to a dipole moment of 22.2 D
for the perpendicular conformation (twisted amino group). For PYRBN, the higher charge
transfer state relevant for the amino rotation (TICT2 state) is lowered only
insubstantially, and does not cross below the lowest *
state even in the fully twisted conformation. Due to the comparable properties (upon
twisting of the alkylamino group) of this state and the TICT state, this second,
energetically higher charge transfer state is referred to as TICT2 state. Induced by the
stabilizing effects of the polar solvent, the S2 excited state is lowered close
to the S1 even for the untwisted (LE for locally excited) conformation of 4BN,
with an almost isoenergetic energy difference of only 0.03 eV. There is an energy barrier
of 0.12 eV against the relaxation along this twisting mode. This relatively high barrier
hints towards a large intensity for the normal fluorescence, with a much smaller charge
transfer band than for PIPBN, due to the higher energy barrier. This predicted
fluorescence behavior is in agreement with experiments [21].
The RICT state, caused by the cyano hybridization mode, is decreased below energy of
the S1 excited state of the unrelaxed vertically excited LE state: Due to the
high dipole moment of this state, This excited state is lowered from the S7
state in gas phase [22] to the S3 by explicit inclusion of solvent effects of
acetonitrile with the SCRF model. The dipole moment varies from 26.0 D for the untwisted
conformation, to 14.7D for the energy minimum at =130o from
there on rising up to 19.8 D for this state with =110o. The
energy barrier against this intramolecular relaxation mode is 0.14eV, more than five times
as much as the energy supplied by thermal effects at room temperature. The lowering of the
dipole moment for cyano bending angles around =120o is a
strong indication against the charge transfer nature of the observed state. Even though
the energy lowering of this charge transfer state is in favor of the RICT hypothesis, the
high energy barrier of 0.14 eV and the low dipole moment reject the RICT hypothesis for
PYRBN.
For PYRBN, the widely discussed solvent induced pseudo Jahn Teller theory is supported
by the minimal energy gap of 0.03 eV between the S1 and the S2
states, permitting vibronic coupling between these two states. Combined with the nitrogen
inversion as a promoting mode, this pseudo Jahn-Teller hypothesis is found to be a
possible explanation for the emission of the red-shifted fluorescence. Even though the
charge transfer state is lowered below the *
state in the untwisted conformation (see Figure 3) for PYRBN, the energy gap of less than
0.03 eV makes this inversion of the states inconsequential, making these two states nearly
isoenergetic.
back to Contents.
3.3. Results for N-piperidinyl-benzonitrile (PIPBN):
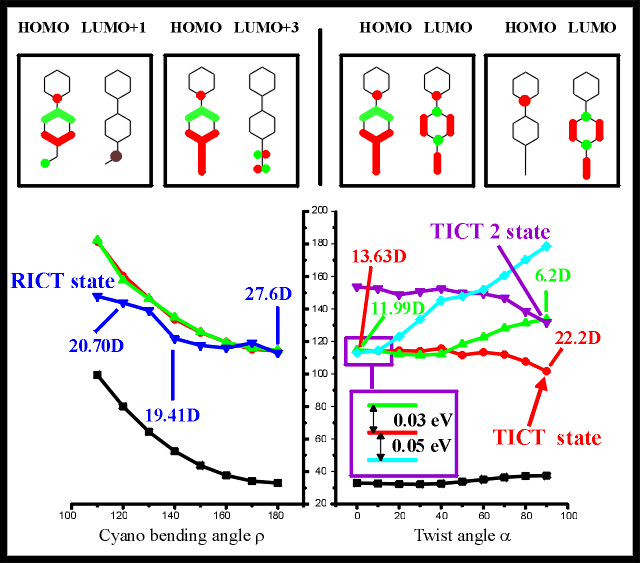
Figure 4: Excited state energies and orbital analyses for PIPBN in acetonitrile
for varying cyano bending angle (bottom left) and for varying amino
twist angle (bottom right). Orbital compositions are presented (top,
from left to right) for the RICT state at =120o and for the
S1 (the charge transfer state becoming the RICT state upon cyano bending) at =180o and for the S3 charge transfer state at =0o and for the same state (TICT state) at =90o
For the energetic minimum of the electronic ground state of PIPBN, the six-membered
alkylamino substituent is found to be in a chair conformation. The excitation energies and
orbital compositions of the charge transfer states considered by the TICT (alkylamino
rotation) and RICT (bending of the cyano group) hypotheses are presented in Figure 4 for
PIPBN. The TICT state, with a twisting angle of 90o, is strongly stabilized
below the S1 state of the vertically excited conformation. As with 4BN, a TICT2
state is lowered energy for large twist angles crosses under the locally excited state for
=90o. The S1 state of B1 symmetry
consists of a benzonitrile * transition.
The S2 state consists of a HOMO/LUMO transition, with a partial electron
transfer from the amino nitrogen lone pair to the benzonitrile moiety. The relevant
orbitals change towards an increasing charge separation for a twist angle of =90o, resulting in a localized HOMO on the amino nitrogen and a
LUMO delocalized over the benzonitrile acceptor group. The charge transfer state crosses
below the lowest * state around =50o. The dipole moment of the * state is lowered from 11.9D to 6.2D for =0o
to =90o respectively. This twisting of the amino group
yields an increase of the dipole moment for the namino*
charge transfer state (TICT state) from 13.6D (=0o up to
22.2D (=90o According to these calculations, there is no
energy barrier against twisting of the alkylamino subunit, strongly favoring the TICT
hypothesis as an explanation for the dual fluorescence of PIPBN.
Due to the high dipole moment of this state, this excited state is lowered from the S8
state found in gas phase calculations [22] below the *
state by explicit inclusion of acetonitrile by means of the SCRF method, making this
state. As the energy differences between this Namino-Ccyano charge
transfer state and the * state and the n* state are only 0.05 eV and 0.03 eV, respectively, these states
are considered to be nearly isoenergetic, making this reversed order of the excited state
negligible. Even though the dipole moment varies only from 27.6 D for the untwisted
conformation, to 19.4 D for =130o this state is not lowered
sufficiently in energy (below the energy of the S1 state of the untwisted
conformation). For larger angles, the dipole moment rises again, to 20.7 D for =110o accompanied by a corresponding increase of the excited
state energy. As for PYRBN, due to the lowering of the dipole moment for cyano bending
angles of =120o this excited state seems to be no charge
transfer state. Contrary to 4BN and PYRBN, the so-called RICT state is not lowered in
energy with increasing cyano beding angles . This effect is due to a
more lowered energy of the vertically excited state, caused by the lowering of another
excited state, causing vibronic coupling of this state with the S1 and the S2
states. In some part this different behaviour of the RICT state of PIPBN can also be
caused by a insufficientactive space (CISD=10), which could not be increased sufficiently
with the used method. The state relevant for the relaxation along the cyano bending mode
(RICT) is not lowered under the energy of the S1 excited state of the
vertically excited conformation, making a relaxation along the RICT mode impossible for
PIPBN.
The calculations in acetonitrile yield the amino-cyano charge transfer state as nearly
isoenergetic with the lowest * state and
the lowest n* state. This other state is lowered due to its
large dipole moment for the vertical excitation conformation. Negligible energy gaps of
only 0.05 eV between the (S1 and the S2) and 0.03 eV (between the S2
and the S3) are small enough to favor vibronic coupling between these states.
As for 4BN and PYRBN, the solvent induced pseudo Jahn-Teller theory is an acceptable
explanation for PIPBN.
back to Contents.
3.4. Comparison to Experimental Data:
The absence of a barrier for the amino twisting mode for PIPBN implies the reduction of
the normal fluorescence band to small intensity in the spectrum of PIPBN. This is found to
be in excellent agreement with experimental data [21], which shows an intense charge
transfer band with only a small shoulder found for the LE emission.
| |
|
AN, calc. |
DE, calc. |
DE, exp. |
4BN |
LE |
357 |
354 |
350 - 360 |
| |
TICT |
548 |
--- |
|
| |
RICT |
592 |
--- |
|
PYRBN |
LE |
355 |
351 |
350 - 360 |
| |
TICT |
540 |
456 |
450 - 460 |
| |
RICT |
606 |
491 |
|
PIPBN |
LE |
355 |
356 |
350 -360 |
| |
TICT |
582 |
476 |
450 - 460 |
| |
RICT |
580 |
434 |
|
For a comparison with experimental data, further excited state calculations with the
explicit inclusion of solvent effects were performed for diethylether as a solvent. For
these calculations in diethylether, only the resulting wavelengths were calculated for the
TICT (=90o RICT (=120o and
LE (=0o =180o state, with no
regard to the energy barrier associated with these relaxation modes. The calculated normal
fluorescence bands of 354 nm, 351 nm and 357 nm for the TICT states of 4BN, PYRBN and
PIPBN, respectively, in diethylether and acetonitrile are in excellent agreement with the
experimental value of 350 - 360 nm in diethylether. As experimental data gave no second
fluorescence for 4BN in diethylether, only the normal fluorescence out of the vertically
excited state is presented for this molecule. For the second, red shifted fluorescence,
the fluorescence bands are predicted in the correct wavelength region for PYRBN and PIPBN.
back to Contents.
4. Conclusion:
Gas phase calculations are a valuable tool for the description of relaxation mechanisms
leading to the emission of a red-shifted fluorescence band, as distinctive charge
separation and a large dipole moment calculated are correlated to the solvent induced
energy lowering of these excited states,. This makes gas phase calculations a valuable
tool for the description of the favored relaxation mechanisms. Nevertheless, the explicit
inclusion of solvent effects is necessary for the correct description of energy barriers
and energetic stabilization caused by solvent effects.
The gas phase calculations presented in a previous paper [22] gave no sufficient energy
lowering of a charge transfer excited state even for large wagging angles . Due to the absence of an energetically lowered WICT state for these N,N-bridged
alkylaminobenzonitriles, the SCRF model was used to describe the solvent induced energy
lowering of the excited states in acetonitrile. As in gas phase, no lowering of a charge
transfer (CT) state below the S1 excited state was observed even for large
wagging angles. Therefore the WICT model, proposing large amino wagging angles and the amino inversion as the promoting mode of the stabilization of a
charge transfer state as, is rejected for this class of compounds.
The RICT hypothesis is rejected for these molecules due to the high barrier against the
cyano bending mode for 4BN and PYRBN, and due to the absence of an energy lowering below
the vertically excited S1 state for PIPBN.
Twisting of the amino group (TICT) is found to be possible for 4BN and unfavorable for
PYRBN. For all molecules, another higher charge transfer (TICT2 state) is significantly
lowered in energy for larger twist angles . The calculations predict
different amino twist mode energy barriers, resulting in varying intensities for the CT
and LE fluorescence bands, which is in accordance with the TICT hypothesis. Even though
the TICT hypothesis has strong merits for this class of compounds, the large intensity of
the charge transfer band for PYRBN can not be explained in a satisfying way by the TICT
model alone.
Due to the small energy differences of 0.04 eV, 0.03 eV and 0.05 eV between the S1 and
the S2 excited states for 4BN, PYRBN and PIPBN, respectively, vibronic coupling, as
proposed by the pseudo Jahn-Teller effect is found as a good description for these N,N-bridged
aminobenzonitriles.
The calculated occurrence dual fluorescence in acetonitrile for 4BN, PYRBN, and PIPBN
is in accordance with experimental data. Calculated fluorescence wavelengths for PYRBN and
PIPBN agree with the experiment, the calculated fluorescence wavelengths for the normal
fluorescence in 4BN, PYRBN and PIPBN are in excellent agreement with experiments.
back to Contents.
5. Acknowledgments:
We thank the Fonds zur Förderung der wissenschaftlichen Forschung,
(P-11880-CHE) in Austria for generous financial support.
back to Contents.
6. References:
- E. Lippert, W. Lüder, F. Moll, W. Nägele, H. Boos, H. Prigge, I. Seibold-Blankenstein,
Angew. Chem. 73, 695 (1961).
- Z. R. Grabowski, K. Rotkiewicz, A. Siemiarczuk, D. J. Cowley, W. Baumann, Nouv. J.
Chim, 3, 443 (1979).
- E. Lippert, W. Rettig, V. Bonacic-Koutecky, F. Heisel and J. A. Miehe, Adv. Chem.
Phys. 68, 1 (1987).
- W. Schuddeboom, S. Jonker, J. Warman, U. Leinhos, W. Kühnle, K. Zachariasse, J.
Phys. Chem. 96, 10809 (1992).
- L. Serrano-Andrés, M. Merchán, B. Roos, R. Lindh, J. Am. Chem. Soc. 117,
3189 (1995).
- K. Rotkiewicz, K. H. Grellmann, Z. R. Grabowski, Chem. Phys. Lett. 98, 315
(1973).
- W. Rettig, V. Bonacic-Koutecky, Chem. Phys. Lett. 62, 115 (1979).
- W. Rettig, Angew. Chem. 98, 969 (1986); Angew. Chem. Int. Ed. Engl. 25,
971 (1986); W. Rettig, Volume 169 of Topics Curr. Chem., Springer Verlag,
Berlin, 1994.
- A. Gorse, M. Pesquer, J. Mol. Struct. (THEOCHEM), 281, 21 (1993).
- A. Gorse, M. Pesquer, J. Phys. Chem, 99, 4039 (1995).
- K. A. Zachariasse, Th. Von der Haar, U. Leinhos, W. Kühnle, J. Inf. Rec. Mat. 21,
501 (1994).
- U. Leinhos, W. Kühnle, K. A. Zachariasse, J. Phys. Chem., 95, 2013
(1991).
- K. A. Zachariasse, T. van der Haar, A. Hebecker, U. Leinhos, W. Kühnle, Pure Appl.
Chem. 65, 1745 (1993).
- K. A. Zachariasse, M. Grobys, T. van der Haar, A. Hebecker, Yu. V. Il\222ichev, O.
Morawski, I. Rückert, W. Kühnle, J. Photochem. Photobiol. A: Chem. 105,
373 (1997).
- A. L. Sobolewski, W. Domcke, Chem. Phys. Lett. 250, 428 (1996).
- A. L. Sobolewski, W. Domcke, Chem. Phys. Lett. 259, 119 (1996).
- A. L. Sobolewski, W. Sudholt, W. Domcke, J. Phys. Chem. in press.
- W. Rettig, J. Phys. Chem. 86, 1970 (1982).
- W. Rettig, Ber. Bunsenges. Phys. Chem. 95, 259 (1991).
- D. Braun, W. Rettig, Chem. Phys. Lett 268, 110 (1997).
- K. A. Zachariasse, M. Grobys, Th. Von der Haar, A. Hebecker, Y. V. Il'ichev, O.
Morawski, I. Rückert, W. Kühnle, J. Photochem. And Photobiol. A: Chemistry 102,
59 (1996).
- R. Schamschule, A. B. J. Parusel, G. Köhler, Internet J. Chem., 1998, 1,
5.
- G. Rauhut, T. Clark, T. Steinke, J. Am. Chem. Soc. 115, 9174 (1993).
- M. Dewar, E. Zobisch, E. Healy, J. Stewart, J. Am. Chem. Soc. 107, 3902
(1985).
- G. Rauhut, A. Alex, J. Chandrasekhar, T. Steinke, W. Sauer, B. Beck, M. Hutter, P.
Gedeck, T. Clark, VAMP6.1, Oxford Molecular Ltd., Oxford, 1997.
back to Contents.
|
